
Even though medicines go through rigorous toxicity testing prior to becoming available on the market, there can be unplanned adverse or side effects once they are used by patients.
Pharmacovigilance is the science and activities relating to the detection and reporting of side effects of a medicine, together with measures to minimise these risks.
You or your child may be treated with one or several medicines. Your experience with taking these medicines is precious: when shared with other patients by reporting them to authorities, it increases the knowledge we have on these medicines, such as how different people react to the same product.
Patients are not just passively taking the treatments that doctors prescribe: they have an active role to play to generate more knowledge about these products. They are vigilant when taking a new medicine, want to be informed of its properties, its effects (both positive and negative) and want this experience to be collected. The below information sets out how you can report adverse events and side effects of a medicine.



-
What is a side effect? What is an adverse drug reaction?
A totally safe and still fully effective medicine does not exist. Even penicillin, which has saved millions of lives can cause severe allergies, which can be deadly.
All drugs have an effect on our body: part of the effect is desired (to prevent, treat or cure a given disease, or ameliorate its symptoms), but part of the effects may be undesired (adverse events). A medicine is authorised to be used in humans when experts estimate that the desired effects on the disease outweigh the undesired effects on your body.
When you do not feel well right after taking a medicine, it may or may not be caused by the medicine. When the effect that you did not desire occurs, it is named an “Adverse Effect”, or “Undesired Effect”. Sometimes it is called a “Side Effect”, “Adverse Event”, or “Undesired Event”, or the term “Suspected Adverse Reaction”. It is only when it is certain that the undesirable effect is due to the medicine that the term “Adverse Drug Reaction” applies. Literally, this means healthcare professionals have established that the undesired effect is a direct reaction to the drug.When experts decide to authorise a medicine, they still wish to learn more about the effect of the medicine as more patients start taking it. When more patients than expected report the same undesired events, and it can be proven that the medicine is the cause, then the experts may revise their decision, adopt measures to diminish the risks when possible, or ultimately decide to withdraw the drug from the market if the effect is severe.
It cannot be guaranteed that all risks are known at the time a medicine first enters the market. It is likely that some risks will only become known after a medicine receives market approval.
For these reasons, rather than talking about the “safety profile” of a medicine, it is more accurate to use the term “toxicity profile” as all drugs come with some degree of risks.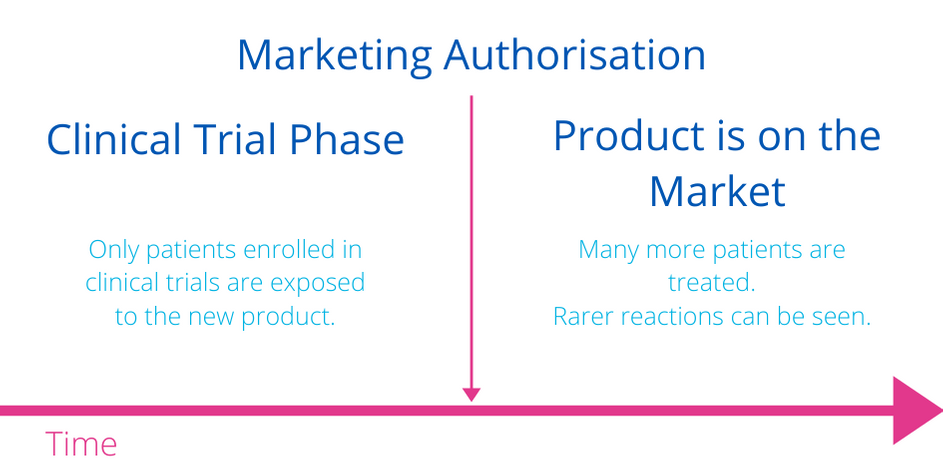
-
What should I do if I suspect an adverse drug reaction?
If the reaction you experience is severe or you fear it could worsen, if you read something about a possible severe reaction in the notice, or if you are anxious for any other reason, always contact your doctor or another healthcare professional. In some cases you may need to contact the emergency services or go to the nearest emergency unit.
What does the inverted black triangle mean?
All medicines are carefully monitored after they are placed on the EU market. If a medicine is labelled with the black triangle , this means that it is being monitored even more intensively than other medicines. This is generally because there is less information available on it than on other medicines, for example because it is new to the market or there is limited data on its long-term use. It does not mean that the medicine is dangerous. The European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) as well as Member States maintains a list of medicines under additional monitoring in the EU, which it updates every month.How to report an adverse event or side effect?
There is a lot you can do, for yourself, and also to help other people who may take the same drug in the future. Your side effect needs to be reported to the persons in charge of drug surveillance: they can analyse what has happened, see if it is due to the drug or not, and propose measures to reduce the risk of it happening to another patient. But first, these experts need to be informed that an adverse drug reaction has occurred.
So your first action will be to talk to your doctor, who will treat the adverse reaction accordingly and then decide to report it or not to a pharmacovigilance expert (when serious, there is an obligation to report).
This report will then be transmitted to EudraVigilance, the European database designed for collecting reports of suspected side effects, maintained by the EMA.
However, healthcare professionals may not always report adverse reactions, due to lack of time, or lack of awareness of the importance to do so.
Or you might prefer to report directly, if for example you feel embarrassed to talk to a healthcare professional about the side effect.
There are different tools for you to report: online forms, mobile applications, printed materials (provided by your doctor, pharmacist or your patient organisations), or by telephone.It is crucial that you fill in all the fields on the reporting form – all the information is important.
See below for how to report an adverse event in your country.
If you need help reporting a side effect in your country you can also phone the national rare disease help line. -
Should I report my error If I make a mistake when taking a medicine?
We can all make mistakes in taking medicines. This may be because the information on the package/ leaflet is unclear or confusing. When this unclear information contributes to an error, such as taking too much of the drug, or too little, or not the right drug, you can report it so as to inform the authorities in charge who can compare the different packages and propose changes.
You can report these mistakes to your doctor or directly to your national competent authority, as listed in the map above. -
Where can I find information on other reports made by patients?
EudraVigilance is the European Medicines’ Agency (EMA) database where all reports from doctors and patients are recorded, and you now have access to these data.
This database contains millions of suspected adverse drug reactions and other information which before April 2012 were accessible only to pharmacovigilance experts and the pharmaceutical industry.
You can access reports made to EudraVigilance database via www.adrreports.eu by searching for a specific medicine.
If you need assistance searching the database contact francois.houyez@eurordis.org.EURORDIS co-authored Patient Reporting in the EU: Analysis of EudraVigilance Data, the largest study to date, by sample size, on patient adverse drug reaction (ADR) reporting that represents the whole EU.
The growing numbers of patient reports indicate patients’ high motivation to report ADRs in the EU and suggests the EU pharmacovigilance legislation has had a positive impact by empowering patients.The EudraVigilance database contained a total of 53,130 patient reports in the 3 years preceding the legislation operation period and 113,371 in the 3 years after.
Patients from the Netherlands, Sweden, Belgium, Austria and Great Britain reported more frequently than in other Member States (numbers of report per million inhabitants). -
What happens after I report an adverse drug effect?
All reports are collected by the pharmacovigilance authorities in your country (public health authorities). They also receive reports from healthcare professionals.
In some cases the pharmacovigilance authorities will need to learn more about what happened, and they can only do so if you accept to provide your contact details with the report. You can also provide the name of your doctor who will then contact you, if needed.
Serious side effects will be processed and analysed in priority. The analysis will be conducted at the level of your country, and your report will also be sent to a European database where reports from all EU countries are stored and analysed.
All information collected at the national level is then sent to EudraVigilance.
EudraVigilance data for centrally authorised medicines (medicines that are evaluated directly by the European Medicines Agency, i.e. all orphan drugs) are analysed on a regular basis, with a two-week or four-week frequency.
There may be some delay between the time patients report and when authorities make adequate decisions. These discussions can take place in your country, and at the European level, so that information, views and decisions can be shared with all Member States.
-
Resources
- The European Medicine Agency’s information on:
- EudraVigilance, the European database of suspected adverse drug reaction reports.
- Safety alerts on medicines will be published by the EMA via their news page and their RSS news feeds.
- List of national competent authorities for reporting adverse events.
-
Pharmacovigilance glossary
A glossary of terms covering topics including adverse reactions and pharmacovigilance.
Adverse event (AE); synonym: Adverse experience
Any untoward medical occurrence in a patient or clinical-trial subject administered a medicinal product and which does not necessarily have to have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign (e.g. an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal product, whether or not it is considered related to the medicinal product
Adverse reaction; synonyms: Adverse drug reaction (ADR), Suspected adverse (drug) reaction
A response to a medicinal product which is noxious and unintended. Response in this context means that a causal relationship between a medicinal product and an adverse event is at least a reasonable possibility.
Adverse reactions may arise from use of the product within or outside the terms of the marketing authorisation or from occupational exposure. Conditions of use outside the marketing authorisation include overdose, misuse, abuse and medication errors.Consumer
A person who is not a healthcare professional – such as a patient, lawyer, friend or relative/parent/child of a patient.
Healthcare professional
For the purposes of reporting suspected adverse reactions, healthcare professionals are defined as medically qualified persons, such as physicians, dentists, pharmacists, nurses and coroners.
Medication Error
Any unintentional error in the prescribing, dispensing, or administration of a medicinal product while in the control of the healthcare professional, patient or consumer.
Name of the medicinal product
The name which may be either an invented name not liable to cause confusion with the common name, or a common or scientific name accompanied by a trade mark or the name of the marketing authorisation holder.
The common name is the international non-proprietary name (INN) recommended by the World Health Organization, or, if one does not exist, the usual common name.
The complete name of the medicinal product is the name of the medicinal product followed by the strength and pharmaceutical form e.g. Elaprase 2 mg/ml concentrate for solution for infusion.Pharmacovigilance
Science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other medicine-related problem.
In line with this general definition, underlying objectives of the applicable EU legislation for pharmacovigilance are:
• preventing harm from adverse reactions in humans arising from the use of authorised medicinal products within or outside the terms of marketing authorisation or from occupational exposure; and
• promoting the safe and effective use of medicinal products, in particular through providing timely information about the safety of medicinal products to patients, healthcare professionals and the public.
Pharmacovigilance is therefore an activity contributing to the protection of patients’ and public health.Serious adverse reaction
Serious adverse reaction means an adverse reaction which results in death, is life-threatening, requires in-patient hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, or is a congenital anomaly/birth defect.
Life-threatening in this context refers to a reaction in which the patient was at risk of death at the time of the reaction; it does not refer to a reaction that hypothetically might have caused death if more severe.
Medical and scientific judgement should be exercised in deciding whether other situations should be considered serious reactions, such as important medical events that might not be immediately life threatening or result in death or hospitalisation but might jeopardise the patient or might require intervention to prevent one of the other outcomes listed above. Examples of such events are intensive treatment in an emergency room or at home for allergic bronchospasm, blood dyscrasias or convulsions that do not result in hospitalisation, or the development of dependency or abuse.
Any suspected transmission via a medicinal product of an infectious agent is also considered a serious adverse reaction.Spontaneous report, synonym: Spontaneous notification
An unsolicited communication by a healthcare professional or consumer to a company, regulatory authority or other organisation (e.g. the World Health Organization, a regional centre, a poison control centre) that describes one or more adverse reactions in a patient who was given one or more medicinal products and that does not derive from a study or any organised data collection scheme.
Stimulated reporting can occur in certain situations, such as direct healthcare professional communication (DHPC), a publication in the press or questioning of healthcare professionals by company representatives, and adverse reaction reports arising from these situations are considered spontaneous reports, provided the report meets the definition above.