Fibrodysplasie ossifiante progressive (FOP)
VIVRE AVEC UNE MALADIE RARE
Manuel et sa famille vivent en Argentine. Il n’avait que 4 ans 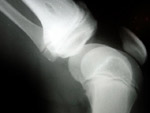 au moment du diagnostic révélant une fibrodysplasie ossifiante progressive, ou maladie de l’homme de pierre, une maladie génétique extrêmement rare qui touche 1 personne sur 2 millions. Ce trouble implique un phénomène rare, à savoir l’ossification des muscles, des tendons, des ligaments et d’autres tissus conjonctifs. Des ramifications osseuses se forment habituellement au niveau des articulations, réduisant progressivement la mobilité du sujet. La fibrodysplasie ossifiante progressive est une maladie qui conduit le corps à produire non seulement un excès osseux, mais un second squelette. Manuel a connu une première poussée en 2000, à l’âge de 3 ans. On parle de poussée à chaque nouvelle formation osseuse, source d’un gonflement des tissus. Le processus est souvent très douloureux. Au départ, les symptômes constatés chez Manuel ont été attribués à une forme de cancer. « Manuel a passé de nombreux tests » explique Moira, sa mère. « Une biopsie a même été pratiquée, alors qu’elle est totalement déconseillée en cas de fibrodysplasie ossifiante progressive, car le choc engendre un excès osseux au point d’impact. On a ensuite diagnostiqué chez Manuel une myofibromatose, puis une fasciite. J’ai commencé à envoyer des centaines de courriels aux hôpitaux du monde entier en décrivant les symptômes observés chez mon fils ! Personne n’avait jamais rien vu de tel, alors nous nous sommes doutés que le diagnostic initial était erroné. L’une des réponses reçues donnait le nom d’un médecin argentin, qui a enfin été capable d’établir le bon diagnostic, en mars 2001. »
au moment du diagnostic révélant une fibrodysplasie ossifiante progressive, ou maladie de l’homme de pierre, une maladie génétique extrêmement rare qui touche 1 personne sur 2 millions. Ce trouble implique un phénomène rare, à savoir l’ossification des muscles, des tendons, des ligaments et d’autres tissus conjonctifs. Des ramifications osseuses se forment habituellement au niveau des articulations, réduisant progressivement la mobilité du sujet. La fibrodysplasie ossifiante progressive est une maladie qui conduit le corps à produire non seulement un excès osseux, mais un second squelette. Manuel a connu une première poussée en 2000, à l’âge de 3 ans. On parle de poussée à chaque nouvelle formation osseuse, source d’un gonflement des tissus. Le processus est souvent très douloureux. Au départ, les symptômes constatés chez Manuel ont été attribués à une forme de cancer. « Manuel a passé de nombreux tests » explique Moira, sa mère. « Une biopsie a même été pratiquée, alors qu’elle est totalement déconseillée en cas de fibrodysplasie ossifiante progressive, car le choc engendre un excès osseux au point d’impact. On a ensuite diagnostiqué chez Manuel une myofibromatose, puis une fasciite. J’ai commencé à envoyer des centaines de courriels aux hôpitaux du monde entier en décrivant les symptômes observés chez mon fils ! Personne n’avait jamais rien vu de tel, alors nous nous sommes doutés que le diagnostic initial était erroné. L’une des réponses reçues donnait le nom d’un médecin argentin, qui a enfin été capable d’établir le bon diagnostic, en mars 2001. »
Comme d’autre s maladies rares voire très rares, la fibrodysplasie ossifiante progressive revêt de nombreux aspects négatifs : un diagnostic retardé sinon erroné, aucun traitement, des connaissances scientifiques limitées et une sensibilisation lacunaire à cette maladie chez les professionnels de santé. Peu de temps après avoir eu connaissance du diagnostic définitif, Moira et son mari ont appris l’existence de l’association internationale dédiée à la fibrodysplasie ossifiante progressive (IFOPA). Moira raconte : « Cette association nous a donné toutes les informations dont nous avions besoin. Nous avions beaucoup de choses à apprendre : comment adapter l’environnement de Manuel, comment parler de la fibrodysplasie avec lui, mais aussi avec notre famille… Quelle chance pour nous que cette communauté extrêmement active ait autant progressé ces 16 dernières années dans la connaissance de la maladie ». Les membres de l’IFOPA sont répartis dans 50 pays différents. Le site Internet de l’association héberge des forums où les internautes peuvent échanger des informations en anglais, en espagnol et en portugais. Moira Liljesthröm est devenue un membre actif de la communauté des patients atteints de la maladie de l’homme de pierre en participant à la création en 2003 d’un groupe latino-américain consacrée à cette pathologie (ALAFOP), soit un réseau d’environ 80 personnes touchées par cette maladie et issues de 10 pays d’Amérique latine. Plus récemment, son mari et elle ont fondé la Fundación FOP en Argentine. Cette fondation, elle aussi dédiée à la
s maladies rares voire très rares, la fibrodysplasie ossifiante progressive revêt de nombreux aspects négatifs : un diagnostic retardé sinon erroné, aucun traitement, des connaissances scientifiques limitées et une sensibilisation lacunaire à cette maladie chez les professionnels de santé. Peu de temps après avoir eu connaissance du diagnostic définitif, Moira et son mari ont appris l’existence de l’association internationale dédiée à la fibrodysplasie ossifiante progressive (IFOPA). Moira raconte : « Cette association nous a donné toutes les informations dont nous avions besoin. Nous avions beaucoup de choses à apprendre : comment adapter l’environnement de Manuel, comment parler de la fibrodysplasie avec lui, mais aussi avec notre famille… Quelle chance pour nous que cette communauté extrêmement active ait autant progressé ces 16 dernières années dans la connaissance de la maladie ». Les membres de l’IFOPA sont répartis dans 50 pays différents. Le site Internet de l’association héberge des forums où les internautes peuvent échanger des informations en anglais, en espagnol et en portugais. Moira Liljesthröm est devenue un membre actif de la communauté des patients atteints de la maladie de l’homme de pierre en participant à la création en 2003 d’un groupe latino-américain consacrée à cette pathologie (ALAFOP), soit un réseau d’environ 80 personnes touchées par cette maladie et issues de 10 pays d’Amérique latine. Plus récemment, son mari et elle ont fondé la Fundación FOP en Argentine. Cette fondation, elle aussi dédiée à la fibrodysplasie ossifiante progressive, mène un projet scientifique sur la situation sociale, médicale et juridique des personnes vivant avec des maladies rares en Argentine. Ce projet est financé par l’Agence scientifique et technologique du gouvernement fédéral argentin. Selon Moira, EURORDIS peut aider la Fundación FOP sur ces travaux : « Nous pourrions vraiment profiter de l’expérience d’Eurordis sur la façon dont conduire des enquêtes comme EurordisCare 1 et EurordisCare 2. La coopération entre les réseaux, les groupes et les associations existants contribuera à inscrire les maladies rares, partout, à l’ordre du jour des questions de santé publique, et à sensibiliser la population. »
fibrodysplasie ossifiante progressive, mène un projet scientifique sur la situation sociale, médicale et juridique des personnes vivant avec des maladies rares en Argentine. Ce projet est financé par l’Agence scientifique et technologique du gouvernement fédéral argentin. Selon Moira, EURORDIS peut aider la Fundación FOP sur ces travaux : « Nous pourrions vraiment profiter de l’expérience d’Eurordis sur la façon dont conduire des enquêtes comme EurordisCare 1 et EurordisCare 2. La coopération entre les réseaux, les groupes et les associations existants contribuera à inscrire les maladies rares, partout, à l’ordre du jour des questions de santé publique, et à sensibiliser la population. »
La localisation, en 2006, par des chercheurs de la faculté de médicine de l’université de Pennsylvanie, du gène en cause donne des raisons d’espérer. Cette localisation et l’identification de la mutation génétique à l’origine de la maladie permettent de cibler précisément les travaux visant à mettre au point des traitements adéquats, pas uniquement pour traiter les symptômes, mais bien la maladie elle-même. Ces recherches ont été partiellement financées par l’IFOPA. Aujourd’hui, Manuel est en CM2 et fréquente une école dans un cadre sûr, pour éviter tout trauma. « Manuel éprouve quelques difficultés à lever les bras et à se pencher, mais ça ne l’empêche pas de jouer et de faire bien d’autres choses encore ! » conclut fièrement sa mère.
Cet article a été publié dans l’édition de juin 2007 de notre lettre électronique.
Auteure : Nathacha Appanah
Photos : toutes les photos © Liljesthröm à l’exception des os © Clara Natoli