Fibrodysplasia Ossificans Progressiva (FOP)
MIT EINER SELTENEN KRANKHEIT LEBEN
Manuel lebt in Argentinie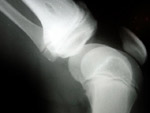 n. Er war erst 4 Jahre alt, als bei ihm eine Fibrodysplasia ossificans progressiva (FOP) diagnostiziert wurde, eine extrem seltene genetische Krankheit, von der nur einer von etwa 1-2 Millionen Menschen betroffen ist. Bei der FOP bildet sich, was sonst ein sehr seltenes Phänomen ist, Knochen in Muskeln, Bänder, Ligamenten, und anderen Strukturen des Bindegewebes. Über die Gelenke hinweg bilden sich in charakteristischer Anordnung Knochenspangen, die zunehmend die Bewegung einschränken. Die FOP ist also eine Krankheit, bei der nicht einfach zuviel Knochen gebildet wird, sondern bei der ein zusätzliches Skelett entsteht. Zum ersten Mal flackerte die Krankheit im Jahr 2000 auf, als Manuel 3 Jahre alt war. In diesem Stadium, wenn der Körper beginnt, neuen Knochen zu bilden, sind die Gewebe angeschwollen und meist auch sehr schmerzhaft. Zuerst dachte man, dass es sich um Zeichen eines bösartigen Tumors handelt. Viele Tests musste Manuel über sich ergehen lassen“, erzählt Manuels Mutter, Moira. Sogar eine Biopsie wurde durchgeführt, was bei FOP ganz und gar unangebracht ist, weil sich an der Stelle des verletzten Gewebes gleich neuer Knochen bildet. Zuerst wurde bei Manuel eine Myofibromatose diagnostiziert und dann eine Fasciitis. Ich begann, an Krankenhäuser in aller Welt e-Mails mit einer Beschreibung der Symptome meines Sohnes zu schreiben, zuletzt waren es 800! Niemand hatte zuvor Ähnliches gesehen, sodass wir annehmen konnten, dass die erste Diagnose falsch war. In einer Antwort auf unsere Mails wurde uns schließlich ein Arzt in Argentinien empfohlen, der dann auch im März 2001 die richtige Diagnose stellte.“
n. Er war erst 4 Jahre alt, als bei ihm eine Fibrodysplasia ossificans progressiva (FOP) diagnostiziert wurde, eine extrem seltene genetische Krankheit, von der nur einer von etwa 1-2 Millionen Menschen betroffen ist. Bei der FOP bildet sich, was sonst ein sehr seltenes Phänomen ist, Knochen in Muskeln, Bänder, Ligamenten, und anderen Strukturen des Bindegewebes. Über die Gelenke hinweg bilden sich in charakteristischer Anordnung Knochenspangen, die zunehmend die Bewegung einschränken. Die FOP ist also eine Krankheit, bei der nicht einfach zuviel Knochen gebildet wird, sondern bei der ein zusätzliches Skelett entsteht. Zum ersten Mal flackerte die Krankheit im Jahr 2000 auf, als Manuel 3 Jahre alt war. In diesem Stadium, wenn der Körper beginnt, neuen Knochen zu bilden, sind die Gewebe angeschwollen und meist auch sehr schmerzhaft. Zuerst dachte man, dass es sich um Zeichen eines bösartigen Tumors handelt. Viele Tests musste Manuel über sich ergehen lassen“, erzählt Manuels Mutter, Moira. Sogar eine Biopsie wurde durchgeführt, was bei FOP ganz und gar unangebracht ist, weil sich an der Stelle des verletzten Gewebes gleich neuer Knochen bildet. Zuerst wurde bei Manuel eine Myofibromatose diagnostiziert und dann eine Fasciitis. Ich begann, an Krankenhäuser in aller Welt e-Mails mit einer Beschreibung der Symptome meines Sohnes zu schreiben, zuletzt waren es 800! Niemand hatte zuvor Ähnliches gesehen, sodass wir annehmen konnten, dass die erste Diagnose falsch war. In einer Antwort auf unsere Mails wurde uns schließlich ein Arzt in Argentinien empfohlen, der dann auch im März 2001 die richtige Diagnose stellte.“
 Die FOP hat mit anderen seltenen und sehr seltenen Krankheiten viel Nachteiliges gemeinsam: Späte und manchmal falsche Diagnose, keine Behandlung, geringe wissenschaftliche Kenntnisse und fehlende Bekanntheit unter den Ärzten. Bald nachdem die Diagnose gestellt worden war, traten Moira und ihr Mann in Verbindung mit der Internationalen Fibrodysplasia-Ossificans-Progressiva-Assoziation (IFOPA). „Der Verband gab uns alle Informationen, die wir benötigten. Wir mussten viele verschiedene Dinge lernen, wie wir für Manuel die Wohnung einrichten, wie wir mit ihm und unserer Familie über FOP sprechen … Wir freuten uns zu sehen, welch große Fortschritte in der Kenntnis der Krankheit diese rege Gemeinschaft in den 16 Jahren ihres Bestehens gemacht hatte“, erklärt Moira. Die IFOPA hat Mitglieder in 50 verschiedenen Ländern, und ihre WebSite bietet Diskussionsforen an, in denen die Menschen Informationen auf Englisch, Spanisch und Portugiesisch austauschen können. Moira Liljesthröm wurde ein aktives Mitglied der FOP-Gemeinschaft und war 2003 an der Gründung einer latein-amerikanischen FOP-Gruppe (ALAFOP) mitbeteiligt. Diese Gruppe umfasst jetzt ein Netzwerk von eta 80 Menschen aus 10 latein-amerikanischen Ländern. In neuerer Zeit haben sie und ihr Mann in Argentinien die Fundación FOP ins Leben gerufen. Diese Stiftung arbeitet über FOP und unterhält ein Forschungsprojekt über die soziale, medizinische und rechtliche Situation von Menschen mit seltenen Krankheiten in Argentinien. Das Projekt wird finanziell von der Agentur für Wissenschaft und Technik bei der Bundesregierung Argentiniens unterstützt. Moira ist überzeugt, dass EURORDIS der Fundación FOP bei diesem Projekt weiterhelfen kann. „Wir können ganz sicher aus der Erfahrung von Eurordis lernen, wie wissenschaftliche Umfragen durchgeführt werden: Beispiele sind die Umfragen EurordisCare 1 und EurordisCare 2. Ich denke, durch die Zusammenarbeit der Netzwerke, Gruppen und Verbände werden wir es schaffen, die seltenen Krankheiten auf die Tagesordnung aller Gesundheitssysteme und ins Bewusstsein der Öffentlichkeit zu bringen.“
Die FOP hat mit anderen seltenen und sehr seltenen Krankheiten viel Nachteiliges gemeinsam: Späte und manchmal falsche Diagnose, keine Behandlung, geringe wissenschaftliche Kenntnisse und fehlende Bekanntheit unter den Ärzten. Bald nachdem die Diagnose gestellt worden war, traten Moira und ihr Mann in Verbindung mit der Internationalen Fibrodysplasia-Ossificans-Progressiva-Assoziation (IFOPA). „Der Verband gab uns alle Informationen, die wir benötigten. Wir mussten viele verschiedene Dinge lernen, wie wir für Manuel die Wohnung einrichten, wie wir mit ihm und unserer Familie über FOP sprechen … Wir freuten uns zu sehen, welch große Fortschritte in der Kenntnis der Krankheit diese rege Gemeinschaft in den 16 Jahren ihres Bestehens gemacht hatte“, erklärt Moira. Die IFOPA hat Mitglieder in 50 verschiedenen Ländern, und ihre WebSite bietet Diskussionsforen an, in denen die Menschen Informationen auf Englisch, Spanisch und Portugiesisch austauschen können. Moira Liljesthröm wurde ein aktives Mitglied der FOP-Gemeinschaft und war 2003 an der Gründung einer latein-amerikanischen FOP-Gruppe (ALAFOP) mitbeteiligt. Diese Gruppe umfasst jetzt ein Netzwerk von eta 80 Menschen aus 10 latein-amerikanischen Ländern. In neuerer Zeit haben sie und ihr Mann in Argentinien die Fundación FOP ins Leben gerufen. Diese Stiftung arbeitet über FOP und unterhält ein Forschungsprojekt über die soziale, medizinische und rechtliche Situation von Menschen mit seltenen Krankheiten in Argentinien. Das Projekt wird finanziell von der Agentur für Wissenschaft und Technik bei der Bundesregierung Argentiniens unterstützt. Moira ist überzeugt, dass EURORDIS der Fundación FOP bei diesem Projekt weiterhelfen kann. „Wir können ganz sicher aus der Erfahrung von Eurordis lernen, wie wissenschaftliche Umfragen durchgeführt werden: Beispiele sind die Umfragen EurordisCare 1 und EurordisCare 2. Ich denke, durch die Zusammenarbeit der Netzwerke, Gruppen und Verbände werden wir es schaffen, die seltenen Krankheiten auf die Tagesordnung aller Gesundheitssysteme und ins Bewusstsein der Öffentlichkeit zu bringen.“ 
Ein Grund, nicht aufzugeben, ist die Entdeckung des FOP-Gens im Jahr 2006 durch Wissenschaftler der University of Pennsylvania School of Medicine. Die Entdeckung des FOP-Gens und der einzigen Mutation, die alle Fälle von FOP verursacht, stellen einen sehr spezifischen Angriffspunkt für mögliche zukünftige Medikamente dar, mit denen nicht nur die Symptome der FOP, sondern auch die Krankheit selbst behandelt werden. Diese Forschungen wurden teilweise durch die IFOPA finanziert. Heute ist Manuel im 5. Grad und besucht eine Schule mit einem sicheren Umfeld, das ihn vor möglichen stumpfen Traumen schützt. „Manuel hat einige Schwierigkeiten, seine Arme zu heben und den Rumpf zu beugen. Das hindert ihn aber nicht, zu spielen und viele an dere Dinge zu tun!“, sagt seine stolze Mutter.
Dieser Beitrag erschien in der Juniausgabe 2007 unseres Rundbriefs.
Autor: Nathacha Appanah
Übersetzer: Ulrich Langenbeck
Fotos: Alle Fotos © Liljesthröm schließen aus Knochen © Clara Natoli